
CNAS+国家CMA二合一扩项现场评审
2022/07/06

演讲主题: 化学药品新注册分类申报资料解读——原料药的特性鉴定
讲师:微谱杂质研究中心分析总监 韩芳霏

简介:韩芳霏,迄今为止已从事药物分析、检测相关工作近10年,尤其在原料药特性鉴定、药品质量研究的工作中积累了丰富的经验。目前已经帮助国内近200家药企完成500多个品种的注册申报工作。熟悉相关法规、指导原则及技术路线,且在技术难点的攻克上具有自己的见解。
各位同仁,大家好。今天的课程主要从申报资料的逻辑出发,与大家分享一下特性药的关键性研究,还有过往工作中遇到的一些经验,希望帮助大家在研发过程中少走弯路。
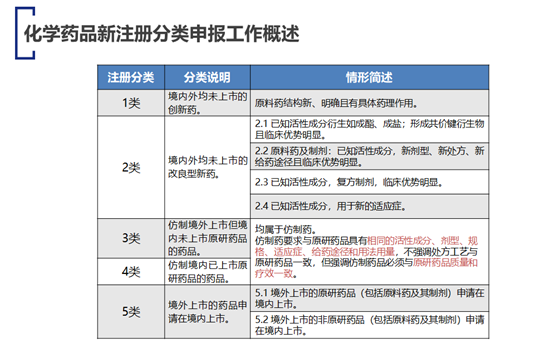
首先,我们看一下化学药品注册分类申报工作的概述。我相信在场各位都比较清楚,在申报化学药品的时候,主要有五个分类,我大概阐述一下。第一类药品是境内外均未上市的创新药。第二类药品是境内外均未上市的改良型新药,是已知活性成分,比如说头孢呋辛钠,它通过人体内的水解过程也会变成有效成分,对于头孢呋辛钠来说,头孢呋辛酯就是一个改良药品。第三类药品是仿制境外上市但境内未上市原研药品的药品。活性成分是已知的,规格、适应症、给药途径和用法用量,不强调处方工艺和原研药品。第四类药品是仿制境内已知上市原研药品的药品,比如说二甲双胍,这个对肿瘤也有抑制作用,如果二甲双胍用于肿瘤治疗的话也是属于改良新药。因此,第三、四类是属于仿制药品。第五类药品是境外上市的药品申请在境内上市。

无论是从申报资料的角度来看,还是从药品研发的角度考虑,对于原料药的研究必定是药品研究过程当中的最重要的步骤。
我们看一下原料药的研究思路,首先要有一个原料生产过程,不管是采购原料药还是自己生成,一般都是通过合成过程,中间会涉及到工艺开发、优化等等。原料药分子得到后会做特性的鉴定。这里面首先要做结构鉴定,判断分子是不是我们想要的结构,另外的理化性质,比如说粒度、溶解度、水分、晶型等有没有满足我们的要求。如果药品当中存在一些杂质,不管是合成当中的有机杂质还是无机杂质都需要做定性的研究。接下来,对于杂质研究的分析方法是不是得到了充分研究和验证呢?我们也会有报告和研究。
原料药的特性鉴定过程当中,我来说几个关键部分。我分了几个类别,这也是我自己的认知。在结构确证方面,我们是解决了产品对不对的问题,表征了产品的分子结构;在晶型研究中,是解决产品是否能够达到充分的生物利用度的问题,表征产品分子的排列方式;在粒度研究中,主要解决产品是否能够表现出合适的溶出速率和含量均匀度的问题,表征产品粒度的宏观大小;杂质鉴定结构定性是定量研究的前提。
在原料药的结构确证上,是参照《化学药物原料药制备和结构确证研究的技术指导原则》,里面一共有两个部分。我们今天重点来看一下结构确证的部分。
如果是骨架结构的的话,可以用单晶X-射线衍射,但是考虑到单晶X-射线衍射的时间成本和人力成本,业内来讲比较常用的方法还是常规方法,通过元素分析或者说高分辨质谱、紫外、红外、核磁等来进行分析。
不同的构型理化性质有不同的差异,或者其氢谱的化学位移和耦合常数是有差异的,还有红外光谱、紫外光谱等,可以通过这些手段进行做表征。另外,分子还会具有手性结构。手性结构的判断一般可以通过手性色谱分离、旋光度测定等方法测定。
在晶型方面,我们主要采用粉末X-射线衍射法,当然也有的用红外、拉曼的方法研究,当然,我们最推荐的还是用X-射线衍射法做研究。
那么是不是所有分子结构都要选择所有的谱呢?是不是中间体结构都不需要去做构型的研究呢?这个没有一个统一的答案,需要根据分子的特性选择合适的测试方法,再加上完整的科学合理完整解析,才会得到这么科学的结论。
接下来,我们讨论一下药物的晶型研究。如果做的创新药或改良型新药就,就要对晶型做完整整的研究。仿制药的晶型研究,一般需要对仿制药中API晶型与原研制剂中API晶型的一致性进行研究。
研究过程中,通常选用X-射线衍射法。我们首先要做一个专属性判断,考察辅料与待研究的API出峰是否有干扰。其次做一个灵敏度和精密度的判断,考察方法是否能够灵敏检出API的出峰,并且在持续的研究中,API的出峰位置是否能够保持良好的精密度。
我们在研究原料药晶型的时候,也要考虑到批间一致性。
最常用的方法是XRD。一般情况下,晶态物质的XRD谱图是呈锐峰的,如果是无定型态物质粉末X射线图会呈弥散峰。判断晶型一致的标准一般有3点:一是衍射峰数量相同;二是两者衍射峰2θ角值位置误差范围在正负0.2°之间;三是相同位置衍射峰的相对峰强度误差在正负5%内。但由于第三点会由于实验技术原因难以实现,所以一般情况下我们考察前2点。
另有一种考察方法是DSC,一般情况下某种晶型的化合物具有特定的熔点和热焓。第三种方法是SEM,可以利用电子显微镜方法进行晶体外形的观察。对于有特定外形的晶型,也可以通过外形来做晶型的判断。
另外我们也收到客户关于晶型杂质控制的发补。一般情况下做XRD晶型定量有三种方法:一种是标准曲线法,适用于两相体系。如果是多相体系,我们推荐的是K值法(外标加入法),这个对于相数没有要求;第三种是全谱拟合法,一般用于科学研究。对于药品申报我们推荐前两种方法。定量研究中特别要关注的是方法的灵敏度。XRD设备的灵敏度一般跟光管强度、检测器灵敏度有关。
接下来,我们讲一下粒度研究。在原料药从溶剂中结晶时,晶体各个生长方向是不一样的,所以晶体就会生长成为不同的外形,比如氯化钠的结晶各个方向的速度是一样的,就会长成立方体的形状。粒度研究主要有三种方式:第一种是显微镜观察,就是直接在光镜下进行粒径的测定;第二种筛分法,这是限度研究的方法;第三种方法是光散射法,是原料药粒度分布研究最常用的方法,相应的仪器设备就是激光粒度仪。
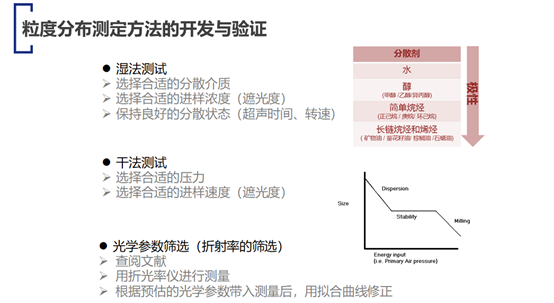
我们根据样品性质不同,可以选择湿法分散和干法分散,如果药品颗粒非常细,这边写了20个微米以下,如果颗粒很细,又比较容易碎的话,推荐湿法测试;如果颗粒较大,不容易碎,推荐干法测试。
前面讲了一些原理或者基础部分,接下来与大家分享一下案例。我们的案例分享也是之前工作的总结。比如说需要将粒度分布订入质量标准,或者是制定合适的主药粒度分布的内控要求。在结构确证中,结构没有提供充分的研究信息,谱图没有做充分的解释说明,都可能造成资料的发补。
粒度研究过程中,预分散的时候,如果您的样品一定要用湿法测定的话,如预分散出现了悬浮、附着、凝聚、团聚、沉淀的情况,可以采用吐温20、吐温80,通过加入表面活性剂来改善样品的悬浮情况。如果在粒度测定过程当中出现了双峰状况,您可以考虑是不是样品本身的分布不均匀,如果没有很好的分布情况就会出现双峰。在数据拟合的时候,可以选择合适的模型;粒度分布均匀的话谱图会呈现正态分布,如果有拖尾的话,考虑样品出现团聚吸附情况,这样就需要选择更合适的分散方式了。
像我们的激光是通过颗粒散射来拟合分布曲线,它测得是不是准确,是不是和实际粒径一样?一定要通过显微镜观察来判断是否符合标准,只有这样才可以说明方法好不好,测得准不准确。后面在做粒度分布的时候一定要注意这一点。
这是结构确证过程中的谱图探讨,我分享一下过往经验中比较异常的谱图,有这样的情况我们一定要进一步思考一下结构和谱图的关联性。
比如核磁测定过程中一维谱图中观测到的样品构象异构的情况,二维谱图中观测到的碳氢耦合相关的异常情况,N原子周围质子和碳信号的特殊峰形等,都需要结合谱图和结构做进一步的思考和探讨。再如在质谱测定过程中如果没有检测到准分子离子峰是否就说明结构有误呢?此时建议大家充分考虑在样品前处理过程中出现的样品水解、酯化等过程,考虑谱图中是否出现水解或酯化产物的出峰。

接下来,跟大家分享一下XRPD实验技术。实验当中如何避免因为实验产生的一些误差?
首先要考虑制样过程,样品表面需平整且与样品台齐平,以避免角度的偏移。其次要注意样品的颗粒尺寸效应和择优取向的问题。如果在谱图中观测到异常的出峰,就要注意是否出现了颗粒尺寸效应和晶体的择优取向问题。
综上,我们总结一下。通过结构确证研究、晶型研究、粒度研究及充分的杂质研究,就可以为原料药研究夯实基础。

扫描上方二维码
关注微谱公众号
电话:400-770-7107
总部:上海市杨浦区国伟路135号9号楼
邮箱:yiyao-marketing@weipugroup.com
Copyright 上海微谱检测科技集团股份有限公司 沪ICP备11022773号 版权声明 Shanghai WEIPU Testing Technology Group Co., LTD. 隐私安全

