
CNAS+国家CMA二合一扩项现场评审
2022/07/06


新冠疫情的持续让 mRNA 疫苗凭借优异的技术优势登上了舞台。mRNA 疫苗将病毒 mRNA 作为抗原导入体内,刺激机体产生免疫反应。与传统疫苗相比,mRNA 疫苗具有安全、高效、易生产的优点,并且以极高的保护率成为所有新冠疫苗种类中热度极高的C位之选。
研究表明 mRNA 5’-cap 结构对于 mRNA 发挥作用是至关重要的,没有 5’-cap 的 mRNA 不能被真核起始因子4E 识别,并且 5’-cap 的存在可以保护 mRNA 5’ 端免受核酸外切酶的降解从而影响 mRNA 的稳定性[1]。因此,检测 mRNA 加帽率毫无疑问成为 mRNA 生产工艺质量分析里面最关键的指标之一。
我国国家药监局药审中心发布的《新型冠状病毒预防用 mRNA 疫苗药学研究技术指导原则(试行)》[2]中明确对 mRNA 加帽率提出要求。客户对于 mRNA 加帽率分析方法的可靠性和完成周期有了更高的期待。本期#微谱药研院·技术共探,我们将一起探讨LC-MS法检测 mRNA 加帽率的原理,以两则案例展开分析,并就常见问题给与答疑。欢迎文末留言讨论。
01:方法原理
首先设计与 mRNA 5’ 端互补的 RNA/DNA 引物探针,探针由两部分序列组成:2’-O-甲基修饰的短 RNA 序列和末端用于引导 RNase H 切割的4-6个DNA 序列。通过梯度退火使引物探针与目标 mRNA 结合,使用链霉亲和素C1磁珠提取目标物,随后利用RNase H在特定的位置进行切割从而将帽子从 mRNA 上酶切下来,最后更换溶剂条件解开引物与 mRNA 5’ 端的结合并回收核苷酸片段,通过LC-MS进行 mRNA 加帽率分析。
02:案例分析
案例一
该分析方法中所用的 RNase H 酶是一种核糖核酸内切酶,能够特异性地降解 RNA-DNA 杂合体的 RNA 链,但 RNase H 酶切割位点的专一性不稳定,除了产生预期的寡核苷酸片段外,可能还会产生其他切割位点的寡核苷酸片段。影响 RNase H 酶切割位点专一性的因素包括探针的长度、RNA中尿嘧啶的修饰等[3]。在这则案例中,除了预期的寡核苷酸片段外,还存在 RNase H 其他位点的切割产物,依赖于质谱的高分辨率,我们也能将其区分开来。
样品信息:
加帽后 mRNA 5’ 序列如下:m7GpppmGGGAGAC***UUGCUUCUG
探针序列:/BioTEG3/mCmCmCmUmCmUm***mGmUmAmAmAmCdGdAdAdGdAdC(其中m代表甲基化,d代表DNA)
单探针设计有可能会产生多个酶切位点,对加帽率的准确性带来一定的挑战。如下文所示

实验步骤:通过退火将 RNA/DNA 引物探针与目标 mRNA 结合,使用 Dynabeads™MyOne™ 链霉亲和素 C1 磁珠提取目标物,再使用 RNase H 酶切并回收核苷酸片段,通过Waters的ACQuity H-class Plus Xevo G2-XS Qtof 搭配ACQUITY Premier Oligonucleotide C18色谱柱对目标 mRNA 进行分析。数据处理软件为UNIFI。
实验结果及分析:图1为样本的 TIC 图,图中最高峰为引物探针峰,因为在前处理过程中为了确保所有 mRNA 都与引物结合,实验加入了过量的引物。相邻峰为样品峰,对其进行解卷积可得到图2的结果。在图2中我们可以明显观察到不同酶切位点的Cap1所对应的峰值,丰度最高的9587.15058对应于酶切位点3所产生的Cap1峰,9281.90878对应于酶切位点2所产生的Cap1峰,9907.00309对应于酶切位点3产生的Cap1峰,不同酶切位点所产生的加帽 mRNA 片段可以使用LC-MS区分开来。
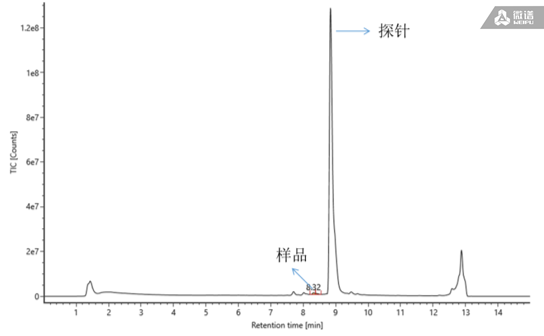
△ 图1 样本TIC图
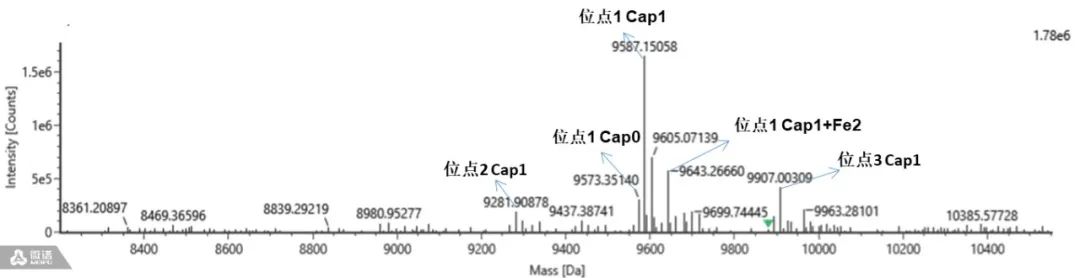
△ 图2 样本解卷积图
mRNA加帽率计算
根据理论分子量匹配对应的Uncapped triphos、Uncapped Diphos、Uncapped Monphos、G Cap、Cap0、Cap1、Cap1 triphos、Cap1 Diphos、Cap1 monphos,并计算其峰面积占比。
Cap1加帽率= Cap1峰面积占比 Cap加帽率= Cap0峰面积占比+ Cap1峰面积占比+ G Cap峰面积占比 对不同酶切位点产生的加帽 mRNA 片段进行加帽率分析,结果如下表:
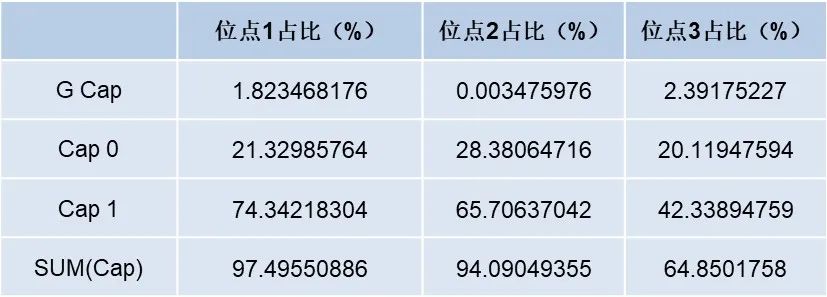
如上表所示,实验产生了至少3个不同酶切位点的数据,不同位点的加帽率比例是有所不同的,因而对实验结果的准确性造成了一定的影响。
因此,对于LC-MS法检测加帽率的实验,可根据目标序列的特点,分别设计合成不同长度的探针序列,介导 RNase H 酶对靶标 mRNA 5’ 端短序列进行切割,从中筛选找到能够产生高比例单一位点切割片段的 RNase H 探针序列,如案例二所示。
案例二
样品信息:mRNA 5’序列如下:m7GpppmGGGAUAAUACUAGUC***GAGAGAAGCCGCCACCAUGUUC一共设计了三种长度的探针,序列如下,其中m代表甲基化,d代表DNA
探针1:/BioTEG3/mCmCmCmUmAmUmUmAmU***mAmAmGmAmCdCdAdGdGdGdG
探针2:/BioTEG3/mCmCmCmUmAmUmUmAm***AmGmAmCmCmAmGmGmGmGdTdGdTdCdTdG
探针3:/BioTEG3/mCmCmCmUmAmUmU***GmUmCmUmCmUmCmUmUmCmGmGmCdGdGdTdGdGdT
检测目的及实验过程:设计不同长度的 RNA/DNA 引物探针与目标 mRNA 序列结合,通过梯度退火使引物探针与目标 mRNA 结合。使用Dynabeads™MyOne™ 链霉亲和素C1磁珠提取目标物,再使用 RNase H 酶切并回收核苷酸片段,最后对目标 mRNA 洗脱,对释放的5'端寡核苷酸进行 mRNA 加帽率分析。使用的分析仪器为 Waters 的ACQuity H-class Plus Xevo G2-XS Qtof,搭ACQUITY Premier Oligonucleotide C18色谱柱。采集的质谱数据通过UNIFI软件进行分析。
实验结果及分析:三种不同长度的探针中产生更高比例单一位点切割片段的为探针1。图3为使用探针1获取的核苷酸片段进样所得的TIC图,保留时间7.92min所对应的峰为样品峰。通过磁珠纯化后的样本比较干净,几乎无杂峰。
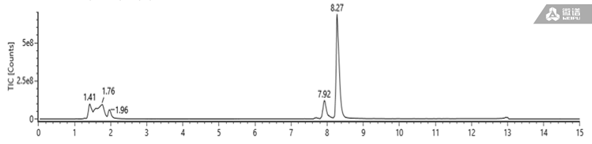
△ 图3 样本TIC图
对质谱数据进行解卷积分析,通过UNIFI 软件进行匹配,匹配结果如图4所示。在加帽反应后,除了产生Cap1外也会生成其他加帽产物,如图4所示的Cap0等。从图中可以观察到使用探针1所产生的酶切位点专一性较高。
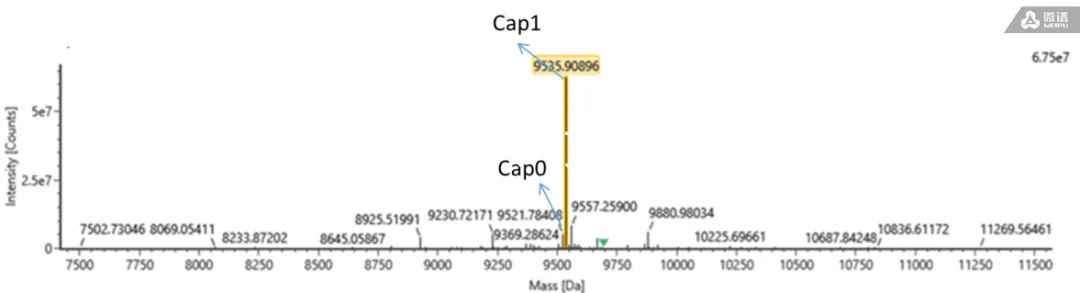
△ 图4 解卷积后谱图
对数据进行加帽率分析,结果如下表:
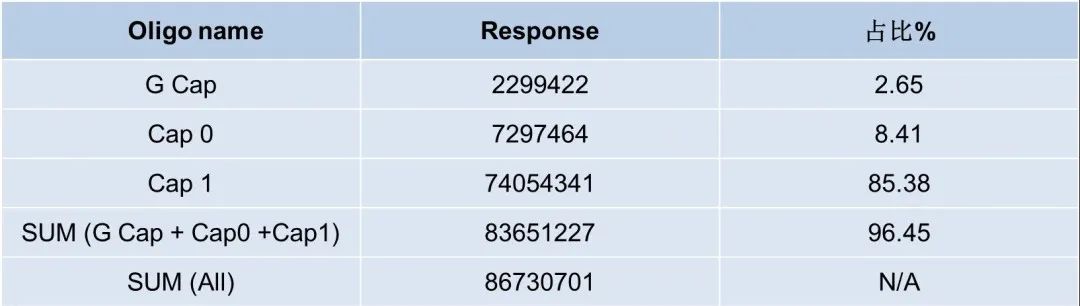
设计合适的引物探针有利于 RNase H 酶切位点的专一性,可增加加帽率数据分析的可信度。在合适探针的辅助下,采用 RNase H 酶切方式获得5’ 端加帽或不加帽的寡核苷酸序列,随后通过LC-MS分离不同大小的寡核苷酸片段,从而评估 mRNA 的加帽率是可行的。
03:常见问答


微谱蛋白药物表征分析平台
实验室配备Agilent及Waters的Q-tof和Thermo Fisher的QE等高分辨质谱,可以满足不同类型生物制品的检测需求。
我们凭借丰富的项目经验,成熟的方法体系,迅捷的方法开发验证和检测周期,目前已助跑多款mRNA产品申报进入决赛阶段。
微谱蛋白药物表征分析平台始于与各大科研院所和高等院校的深度合作。依托微谱生物医药板块强大的仪器配置,完备的质量体系及多年积累的糖库,我们已经搭建出符合国际质量标准的生物制品研发分析与实验室服务平台,可以为客户提供专业的一体化生物制品质量研究服务。
蛋白多肽类药物结构表征:质量肽图、糖谱分析、高级结构、理化分析、产品变异性、生物学活性、产品相关杂质、工艺相关杂质;
mRNA药物结构及关键质量属性:加帽率、Poly A 长度、金属残留、溶剂残留、蛋白酶残留、mRNA鉴别/含量/纯度/杂质、脂质鉴别/含量/纯度/杂质;

扫描上方二维码
关注微谱公众号
电话:400-770-7107
总部:上海市杨浦区国伟路135号9号楼
邮箱:yiyao-marketing@weipugroup.com
Copyright 上海微谱检测科技集团股份有限公司 沪ICP备11022773号 版权声明 Shanghai WEIPU Testing Technology Group Co., LTD. 隐私安全

