
CNAS+国家CMA二合一扩项现场评审
2022/07/06

5月25日,由火石数链×微谱联合推出,微谱药研院医疗器械专场课程,微谱医疗器械安全性研究专家赵中瑞老师作为特邀分享嘉宾的线上演讲——《医疗器械药物吸附&可沥滤物研究流程与案例分享》圆满举行。
本期推文为您带来演讲精彩回顾。同时,为回应直播中行业同仁们的热情提问,我们还特别邀请赵老师返场,附赠更多Q&A福利,欢迎前排围观,留言互动!


医疗器械药物吸附&可沥滤物研究流程与案例分享

赵中瑞
微谱医疗器械安全性研究专家
个人简介:熟稔医疗器械研究相关法规和技术标准,聚焦于输注器具、骨科植入物、血液透析器、雾化器、药械组合等产品的安全性研究工作,在药物相容性研究、金属离子析出研究、化学表征研究、含药医疗器械药物定性定量与释放研究等领域拥有丰富的实践经验。擅长综合运用各方面专业知识,精准解决医疗器械安全性研究中的复杂难题。作为核心技术专家,已经推动了多种医疗器械产品在国内外,包括NMPA、FDA、EU等市场的注册获批。
演讲回放
一、药物相容性概述
药物相容性研究主要是为了考察药品和它的接触材料之间是否会发生浸出或吸附现象。因为这两个作用都可能会导致药品发生一系列质量安全性风险问题。
在药品上市后,用药阶段通常需要通过一些医疗器械将药物注入人体内。输注器具药物相容性它的研究对象主要是输液类医疗器械和预充式医疗器械等。对这类产品而言,药物相容性研究主要包括输注器具对药物的吸附作用和输注器具中某些添加剂、单体等可沥滤物的迁移进入药液两个作用。
展开来讲,为了保障药液输注过程中药物活性成分的有效性,需要进行药物吸附研究和药物质量变化研究;为了医疗器械使用过程中的安全性,还需要展开可沥滤物研究或可浸提物研究等。从产品出发,风险评估是我们进行相容性研究与方案设计的前提和基础。
二、药物吸附研究
进行药物吸附研究,主要是为了保障药液输注过程中药物活性成分的有效性。这主要包括两部分内容:第一,通过药物浓度的变化来表征吸附率,参考法规是YY/T 1550.1 ;第二,对药液进行药物质量变化的研究,这时我们需要结合药物在《中国药典》收录页中的要求的一些质量指标。

药物吸附研究,主要参考标准为YY/T 1550.1-2017。吸附研究一般通过测定输液液体中药物浓度的变化来表征药效的变化。需要注意的是,吸附率是动态变化的过程。
同时,在进行药物吸附研究方案和实验的设计时,可以参考2012年发布的《一次性使用避光输液器产品注册技术审查指导原则》。该指导原则为我们的研究工作提供了一些非常重要的指导。
总体来讲,可以分为五个主要步骤:
01. 选择典型性样品和药物(临床使用规定;药物性质);
↓
02. 结合产品说明书设计实验参数(稀释剂、浓度、流速、输注剂量、温度和时间点等);
↓
03. 制备样品溶液进行方法学验证;
↓
04. 测试各时间点的吸附率和质量变化(酸碱度、不溶性微粒等);
↓
05. 对实验结果进行综合性评价得出结论。
总的来说,输液器具对药物的吸附作用会直接影响药物的治疗效果,同时对于低剂量给药和精确给药的治疗,吸附和解吸附作用,易导致潜在风险,所以科学合理的研究资料可以更好的指导临床用药。作为输液器具产品申报资料的重要内容,药物吸附研究不容忽视。
三、可沥滤物研究
可沥滤物是指医疗器械或材料在临床使用过程中释放出来的物质的统称,一般包括灭菌残留剂、工艺残留物、降解产物以及材料中的单体及添加剂(包括稳定剂、抗氧化剂、增塑剂、着色剂等)等。我们把可沥滤物分成两大块内容:已知可沥滤物和未知可沥滤物。今天的主题主要涉及已知可沥滤物研究方面。
医疗器械的可沥滤物(Leachables)是医疗器械或材料在临床使用过程中释放出的物质的统称。一般包括灭菌残留剂、工艺残留物、材料中的单体及添加剂(包括稳定剂、抗氧化剂、增塑剂、着色剂等)以及降解产物。
以高分子医疗器械为例,其在医疗器械发挥作用的过程中,原材料所用的各种包括增塑剂、抗氧化剂、稳定剂等在内的添加剂,以及生产加工过程中引入的工艺残留均可能直接或通过相关介质进入人体,形成可沥滤物的安全性风险。主要的参考标准是YY/T 1550.2-2019和《医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则-2019》。

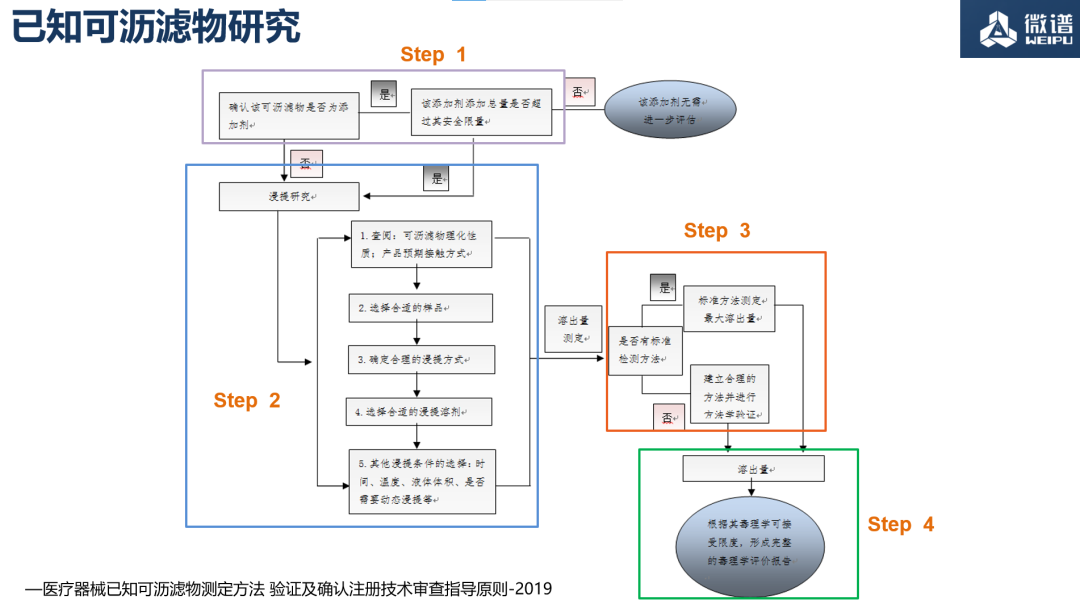
医疗器械已知可沥滤物的研究主要可以分为以下四个步骤:
01. 通过添加量是否超过安全限量(即AL值允许限量 参考GB/T 16886.18)确定目标物;
↓
02. 设计科学合理的浸提实验;
↓
03. 选择合适的仪器,进行测试的方法学研究;
↓
04. 对目标物的溶出量进行安全性评估。
四、案例分享
在案例分享环节,主要介绍了一次性使用避光滴输液器的已知可沥滤物研究与吸附研究;以及一次性使用血液灌流器的已知可沥滤物研究。
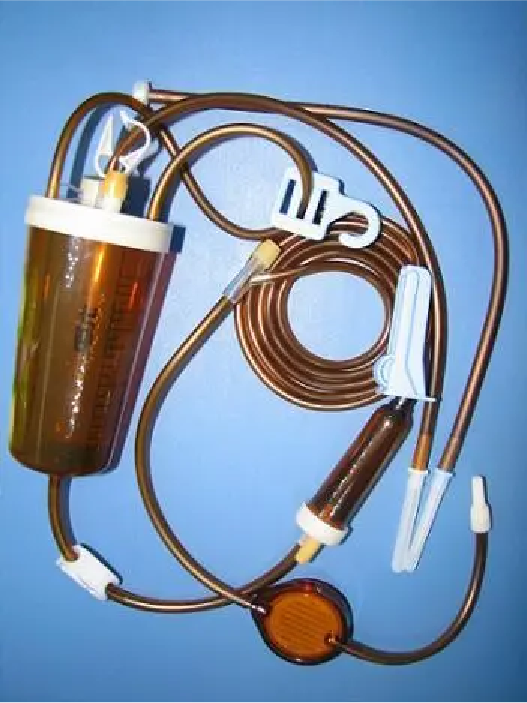
▲一次性使用避光滴输液器
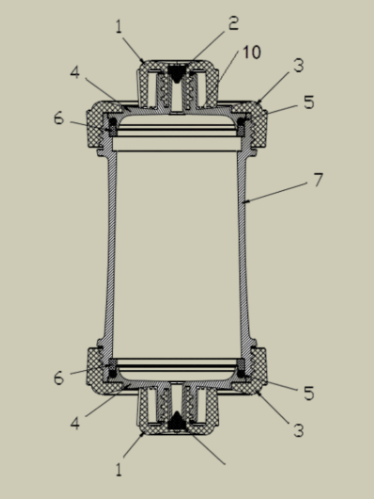
▲ 一次性使用血液灌流器产品

扫描上方二维码
关注微谱公众号
电话:400-770-7107
总部:上海市杨浦区国伟路135号9号楼
邮箱:yiyao-marketing@weipugroup.com
Copyright 上海微谱检测科技集团股份有限公司 沪ICP备11022773号 版权声明 Shanghai WEIPU Testing Technology Group Co., LTD. 隐私安全

